Computational Insights into Cholesteryl Carbamate Derivatives: A Density Functional Theory and Molecular Docking Approach
Anitha Bujji, J. Jeetha Raj, Annapurna Padmavathi Devarakonda*
DOI: DOI: 10.22607/IJACS.2025.1302004
Volume 13, Issue 2 | Pages: 85-90
Abstract
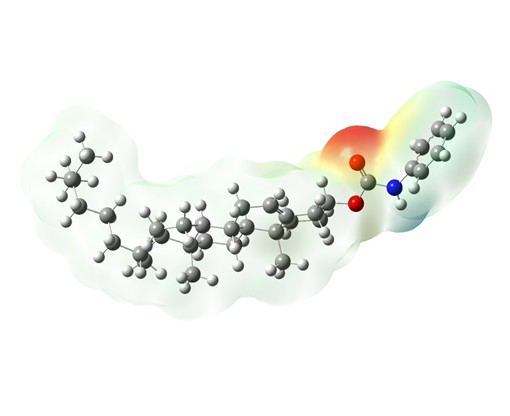
In the study, a series of novel cholesterol carbamate derivatives were computationally investigated using Density Functional Theory (DFT) and Molecular Docking methods to evaluate their global reactivity and potential biological interactions. Ten derivatives, incorporating aliphatic and aromatic amide functionalities were modelled and optimised using the B3LYP/6-31G level of theory. Key global description such as HOMO-LUMO energy gap (ΔE), chemical hardness (η), softness (S), electronegativity (χ) and global electrophilicity index (ω) were calculated to assess the stability and reactivity of each compound. Among them, the 4-Nitroaniline derivative (C8) displayed the lowest energy gap (ΔE =2.68 eV), highest electrophilicity index (ω= 5.5140), and highest dipole moment (8.73 D), suggesting strong reactivity and a potential for favourable interactions with biological targets. Furthermore, molecular docking studies against calf thymus DNA (ct-DNA) revealed that C8 exhibited the most favourable binding, with a binding energy of -7.90 kcal/mole and an inhibition constant of 1.61 μM. Compounds C4 and C7 also showed promising interactions, supported by their moderate energy gaps and high electron donating capacities. The combined computational approach offers valuable insights into the electronic features and the binding potential of cholesteryl carbamate derivatives, paving the way for future biological and experimental validations.
Keywords
Cholesteryl carbamate derivatives Density Functional Theory (DFT) Molecular Docking Binding energy Bioactive compoundsReferences
No references available for this article.
Citation
Anitha Bujji, J. Jeetha Raj, Annapurna Padmavathi Devarakonda*. Computational Insights into Cholesteryl Carbamate Derivatives: A Density Functional Theory and Molecular Docking Approach. Indian J. Adv. Chem. Sci. 2025; 13(2): 85-90.