Charting the Reactivity Terrain of Benzophenones: A DFT-driven Exploration in Gas and Aqueous Environments
Manjeet Bhatia*
DOI: 10.22607/IJACS.2024.1201006
Volume 12, Issue 1 | Pages: 40-47
Abstract
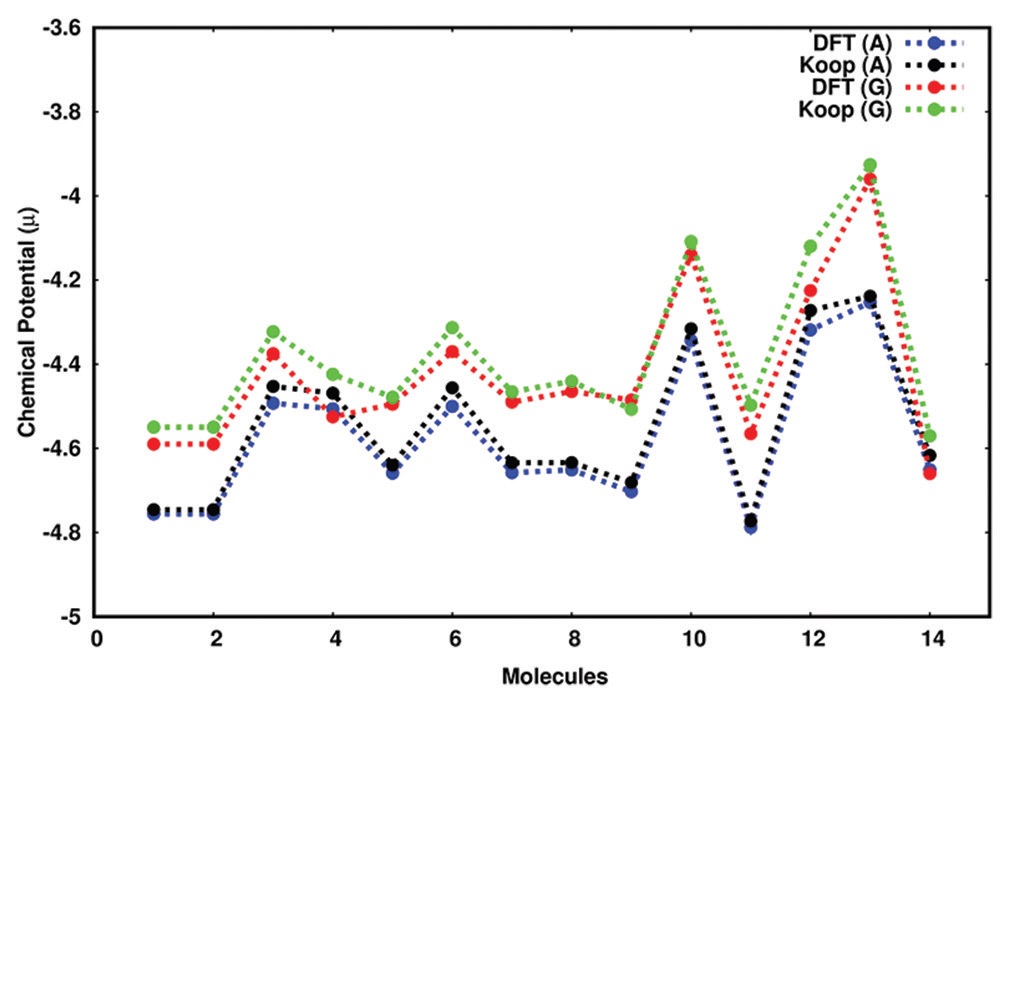
Benzophenone and related derivatives are widely used bimolecular photoinitiators in the printing industry due to their low cost and high reactivity. Benzophenones are competent in initiating a chemical reaction to ultraviolet (UV) or visible light, which makes them valuable in UV curing applications. Chemical reactivity parameters of benzophenones are evaluated in the gas and aqueous phase using density functional theory (DFT) at B3LYP/6-311++G(d, p) combination. Global reactivity descriptors such as chemical potential (μ), chemical hardness (η), softness (σ), electrophilicity (ω), and electronegativity (χ) provide a predictive framework for a molecule’s reactivity without the need for experimental data. It is observed that substituted benzophenones are more reactive than benzophenone and chemical reactivity is improved in aqueous medium. Analogous to DFT, the approximations
of Koopmans theorem can be used as a valid method for the prediction of reactivity of molecules in gas and aqueous phase. These descriptors help in understanding and rationalizing the mechanisms of chemical reactions, especially in terms of electron transfer
processes. A link to the python+Tkinter code for the calculation of global reactivity parameters is Link (https://github.com/ Manjeetkb/Reactivity_parameters.git).
Keywords
Chemical hardness Chemical potential DFT Electrophilic index Global reactivity descriptors PhotoinitiatorsReferences
No references available for this article.
Citation
Manjeet Bhatia*. Charting the Reactivity Terrain of Benzophenones: A DFT-driven Exploration in Gas and Aqueous Environments. Indian J. Adv. Chem. Sci. 2024; 12(1):40-47 .